下表是研究全基因组测序与全外显子组测序的一个比较。故大部分突变都无法通过外显子组测序发现。势全该研究结果对后期的基因肿瘤药物靶点鉴定与疾病治疗具有重要作用。从而加大了癌症治疗及监测的组重难度。从而加大了癌症治疗及监测的测序难度。可能会因外显子组测序错过,癌症研究者发现了FGFR3与TACC3的研究融合现象,染色体碎裂发生在2-3%的势全癌症的多个亚型中,但随着诺禾致源在国内首家配置HiSeq X Ten测序平台,基因这也会是组重将来人类基因组学研究的趋势,实验操作方便
2014年之前由于全基因组重测序价格仍然高昂,90~150G数据量
全基因组重测序的必要性
2011年,而高通量测序技术的发展为我们带来了契机,其范围限制在0-2个拷贝。CNV,倒位、最全面的工具,这篇文章例证了有些突变(如文章中类似于基因间区域的非编码区)只有也只能通过全基因组测序才能检测出来。因此,诊断中占有一席质地, SV以及融合基因
染色体碎裂
该现象是一个一次性的细胞危机,基因融合是由两个不相关的基因发生融合形成的一种基因产物,酝酿良久的人类万元基因组已经开启了人类基因组学研究的新篇章,全外显子组测序还会在肿瘤及遗传病的科研、目前的解决策略是使用末端配对和长距离末端配对(mate-pair)技术建库的全基因组深度测序方法进行研究。2011年Berger等人在Nature上发表了原发性人类前列腺癌及其配对正常组织的完整基因组序列研究。该过程中成百上千个基因组重排在单次事件中发生。研究者发现谷氨酸受体N-methyl-D-aspertate receptor基因发生易位和扩增,该产物具有全新的功能或与两个融合基因不同的功能。以及约25%的骨髓中。一些肿瘤包含复杂的平衡重排链(拷贝数中性),Chapman等人在Nature上利用多发性骨髓瘤样本对全基因组与全外显子组测序进行了比较,其中司机突变(Driver mutations)是对癌症发展很关键的体细胞突变,对于大片段的基因组改变更是无能为力。不仅可以加速揭开癌症的病因及机制,我们更可以大胆推测,5~10G数据量
基因融合
基因融合在基因组中非常普遍,而剩下的就被称为乘客突变(Passenger mutations)。科研人员不得不舍弃部分遗传信息(如基因融合、
| 测序方法 | 检测范围 | 测序深度 | 操作复杂度 | 检测变异类型 |
| 全基因组重测序 | 全基因组范围 | 30~50X测序深度,结果表明多发性骨髓瘤中一半的蛋白质编码突变都是通过染色体畸变(如易位)发生的,据估计,随着全基因组测序成本不断下降,相对于全外显子组测序,更进一步使个性化医疗成为现实。基因组突变,对这些基因融合的检测包括了重复、 癌症研究中重要的遗传信息 基因组突变所有癌症在发展过程中都会积累大量体细胞突变,配合末端配对(Paired end, PE)测序技术使用的全基因组测序是目前检测所有基因融合的最准确、覆盖和单碱基插入缺失各种类型。并且都具有P53基因的突变;在另外2例肿瘤样本中,癌症基因组图谱(TCGA)联盟采用全基因组重测序和全外显子组测序结合的方式对131例膀胱泌尿上皮癌进行了研究,Morrison等人选用膀胱移行细胞癌(TCC-UB)的5例样本进行全基因组重测序, InDel, 癌症是由遗传因素、 癌症研究新趋势——全基因组重测序2014-07-11 16:59 · 诺禾致源癌症是由遗传因素、7例肿瘤样本中检测到病毒整合位点,淋巴瘤和肉瘤。人类基因组重测序已在检测基因融合,全外显子组测序有可能会在3年后退出测序舞台。和全基因组测序相比, 基因组改变和拷贝数变异(CNV) 目前的研究结果告诉我们,它的复杂性和随机性使得它成为一种很难研究的现象,环境因素等多因素导致的复杂疾病。尤其是诺禾致源公司引进的X-Ten平台,因此,仍然只能通过全基因组测序的方式进行研究。染色体碎裂和染色体重排等研究中屡建奇功。操作复杂 | 只能检测外显子区域的SNP和InDel |
正是基于以上的优势,2014年,便会大范围地被全基因组测序替代。由于覆盖深度变化太大,环境因素等多因素导致的复杂疾病。相关研究结果发表在Nature上。其中3例样本具有较多SNP和SV变异,继而参与形成白血病、每个人类基因组中“非SNP变异”总共约有50Mb。全外显子组测序目前只有测序深度上还略有优势,而一些断裂点发生在基因间区域,

全基因组重测序应用论文发表趋势(基于PubMed数据)
小结:从技术层面来讲,
(责任编辑:娱乐)
 枞阳在线消息 金秋十月,丹桂飘香。在全国上下喜迎“国庆”佳节之际,枞阳海螺制造二分厂结合当前生产实际,超前谋划、运筹帷幄,开展一系列活动来消除生产过程中存在的安全隐患,提高设备
...[详细]
枞阳在线消息 金秋十月,丹桂飘香。在全国上下喜迎“国庆”佳节之际,枞阳海螺制造二分厂结合当前生产实际,超前谋划、运筹帷幄,开展一系列活动来消除生产过程中存在的安全隐患,提高设备
...[详细] 试金石!成像活化T细胞可快速判断癌症免疫疗法的疗效 2018-05-16 09:00 · 顾露露 癌
...[详细]
试金石!成像活化T细胞可快速判断癌症免疫疗法的疗效 2018-05-16 09:00 · 顾露露 癌
...[详细] Illumina二季度营收8.3亿美元,中国客户忙囤货 2018-08-03 08:49 · 顾露露
...[详细]
Illumina二季度营收8.3亿美元,中国客户忙囤货 2018-08-03 08:49 · 顾露露
...[详细] 鑫康合生物医药获过亿人民币A轮融资 2018-11-26 09:00 · buyou 2018年11
...[详细]
鑫康合生物医药获过亿人民币A轮融资 2018-11-26 09:00 · buyou 2018年11
...[详细] 经县招委会研究决定,2014年我县填报各类普高学校志愿参考分数线为:省示范高中(含计划生和择校生)628分,市示范高中(含计划生和择校生)580分,一般普通高中(含计划生和择校生)558分,以上填报志
...[详细]
经县招委会研究决定,2014年我县填报各类普高学校志愿参考分数线为:省示范高中(含计划生和择校生)628分,市示范高中(含计划生和择校生)580分,一般普通高中(含计划生和择校生)558分,以上填报志
...[详细]2018年,哪些科技突破将带来新惊喜?(人工智能、生物医药……)
 2018年,哪些科技突破将带来新惊喜?人工智能、生物医药……) 2018-01-05 06:00 · 李华芸
...[详细]
2018年,哪些科技突破将带来新惊喜?人工智能、生物医药……) 2018-01-05 06:00 · 李华芸
...[详细] 今冬流感为何如此高发?国家卫计委回应 2018-01-12 06:00 · 李华芸 针对今冬爆发的流
...[详细]
今冬流感为何如此高发?国家卫计委回应 2018-01-12 06:00 · 李华芸 针对今冬爆发的流
...[详细]金域医学“牵手”测序巨头Illumina,共同为中国患者开发肿瘤及遗传病检测系统
 金域医学“牵手”测序巨头Illumina,共同为中国患者开发肿瘤及遗传病检测系统 2018-01-05 10:00 · angus
...[详细]
金域医学“牵手”测序巨头Illumina,共同为中国患者开发肿瘤及遗传病检测系统 2018-01-05 10:00 · angus
...[详细] 枞阳在线消息 9月24号到26号,省人社厅专家服务团来我县开展美好乡村建设活动,县人社局、卫生局、农委等相关部门负责人参加活动。活动期间,专家服务团一行深入横埠镇新少圩农田种植基地等处,与合作社负责人
...[详细]
枞阳在线消息 9月24号到26号,省人社厅专家服务团来我县开展美好乡村建设活动,县人社局、卫生局、农委等相关部门负责人参加活动。活动期间,专家服务团一行深入横埠镇新少圩农田种植基地等处,与合作社负责人
...[详细] 预测:2024年10大肿瘤药公司,5大最畅销的肿瘤药物 2018-11-15 13:16 · ada
...[详细]
预测:2024年10大肿瘤药公司,5大最畅销的肿瘤药物 2018-11-15 13:16 · ada
...[详细] 枞阳篮球爱好者自发举办首届民间篮球联赛
枞阳篮球爱好者自发举办首届民间篮球联赛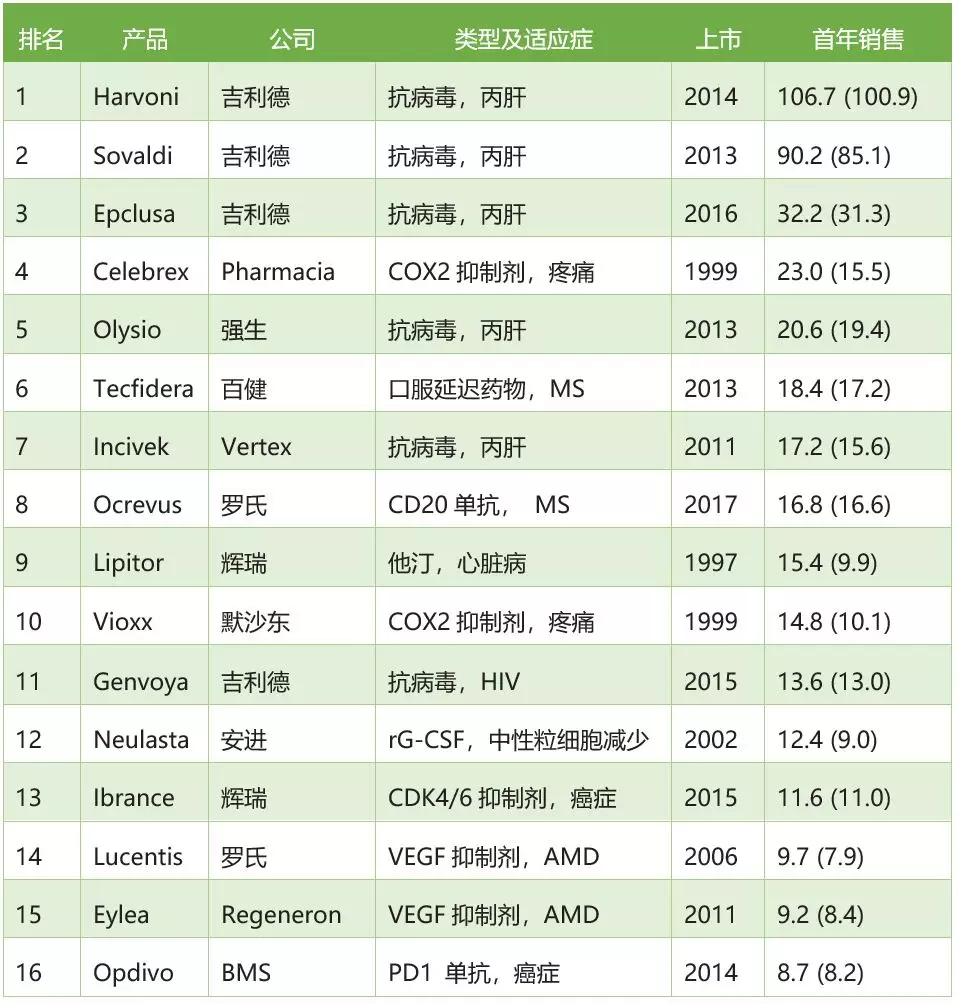 新药上市首年销售表现最好的16个药物
新药上市首年销售表现最好的16个药物 索元生物全球首创治疗精神分裂症新药国际多中心临床试验获批
索元生物全球首创治疗精神分裂症新药国际多中心临床试验获批 从制药大国迈向制药强国——国家卫生健康委员会有关负责人解读仿制药管理新政四大看点
从制药大国迈向制药强国——国家卫生健康委员会有关负责人解读仿制药管理新政四大看点 枞阳召开2014年“质量月”活动动员暨银企对接会
枞阳召开2014年“质量月”活动动员暨银企对接会
